Introduction
Systemic lupus erythematosus (SLE) in the elderly, also called late-onset SLE, is an autoimmune disease that manifests after age 50 years. The clinical course and clinical manifestations differ from those of classic SLE which predominantly affects younger women. Late-onset SLE has a longer lag time to diagnosis and lower rates of immunological disorder than classic SLE. Herein, we report the case of a geriatric man who was diagnosed with SLE at age 60 years with a past history of protein-losing enteropathy (PLE) and vitiligo since age 51 years.
Case
A 60-year-old man was referred to our hospital with a 2-month history of dyspnea, cough, hemoptysis, and lower-limb edema. He had no complaints or history of Raynaud phenomenon, myalgia, and arthralgia. Family history included late-onset SLE in the patient’s father. At age 51 years, he experienced edema, pleural effusion, and ascites, for which he visited another hospital. At the same age, several months after onset of these symptoms, he experienced vitiligo. Renal functions including the blood urea nitrogen (BUN) and serum creatinine (Cr) were normal. Serum total protein (TP) levels were low. Positive results were obtained for antinuclear antibody (ANA), anti-SSA antibody, and anti-RNP antibody. Negative results were obtained for proteinuria, hematuria, and anti-dsDNA antibody. A thoracentesis yielded low TP levels and negative results for the Rivalta test; therefore, the pleural effusion proved to be transudative. Considering the low TP levels, a diagnosis of PLE was made. Therefore, the pleural effusion and ascites were thought to be caused by PLE-induced hypoproteinemia and hypoalbuminemia. At that time, the patient had no positive criteria for the diagnosis of SLE according to the 1997 American College of Rheumatology revised criteria for the classification of SLE (revised ACR criteria) [1]. For PLE, he was treated with an albumin (Alb) infusion once a month in combination with furosemide, followed by furosemide monotherapy. The pleural effusion, ascites, and lower-limb edema nearly resolved, with Alb remaining within the range of 2.2–2.5 g/dL.
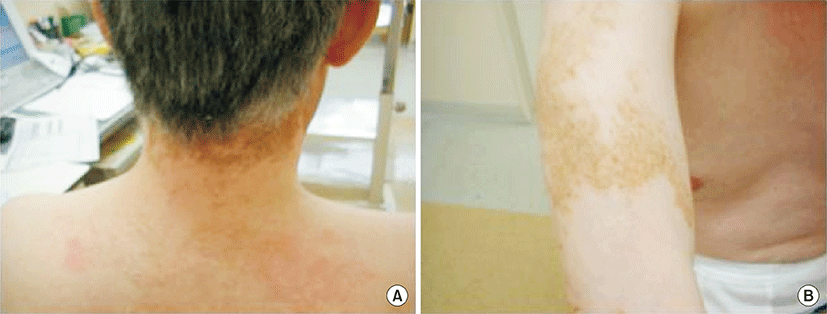
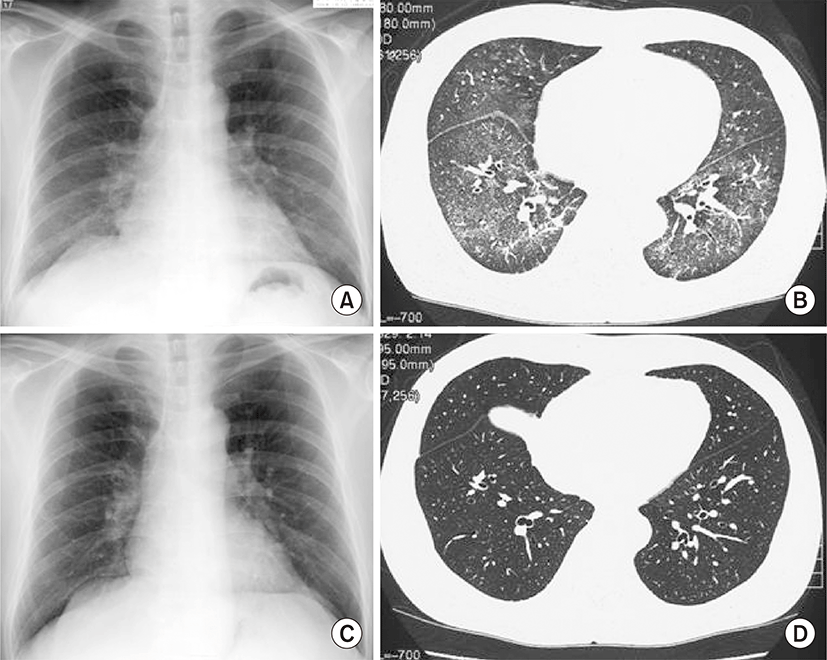
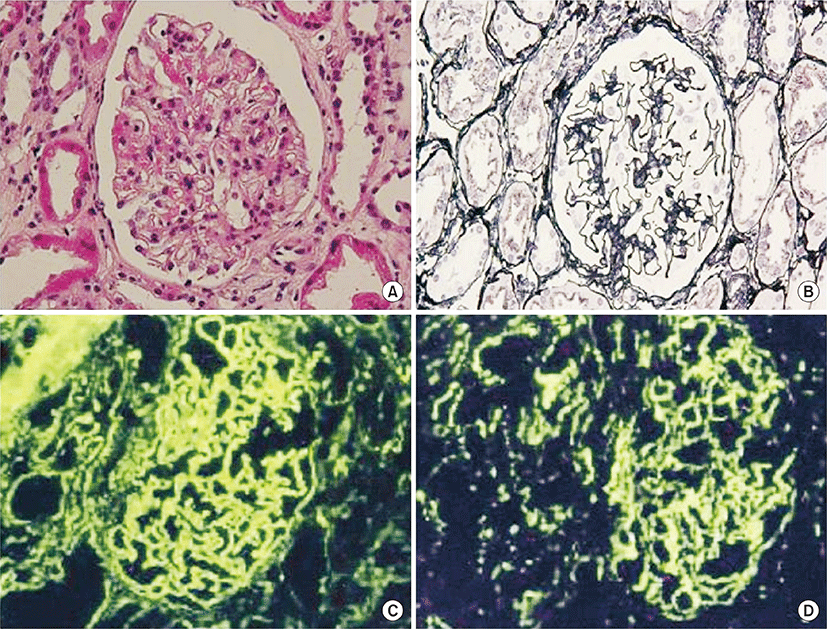
On this visit to our hospital, physical findings were as follows: blood pressure, 136/82 mmHg; heart rate, 86 beats/min; respiratory rate, 25 breaths/min; and body temperature, 38.6°C. Coarse crackles were audible bilaterally, mostly in the lower chest wall. Bilateral lower-limb edema and vitiligo were observed (Fig. 1). Neither muscle tenderness nor joint swellings were recognized. The laboratory findings were as follows: white blood cell count, 7,790/μL (neutrophils, 75.0%; eosinophils, 0%; monocytes, 1.3%; lymphocytes, 23.6%); red blood cell count, 349×104/μL; hemoglobin (Hb), 8.3 g/dL; platelet count, 10.8×104/μL; aspartate aminotransferase, 43 international units (IU)/L; alanine aminotransferase, 18 IU/L; BUN, 15.0 mg/dL; Cr, 0.61 mg/dL; TP, 6.6 g/dL (γ-globulin, 25.3%; normal range, 11.5% to 22.4%); Alb, 1.6 g/dL (normal range, >4.0 g/dL); and C-reactive protein, 1.74 mg/dL (normal range, <0.3 mg/dL). Serological tests showed: immunoglobulin G (IgG), 3,690 mg/dL (normal range, 870 to 1,700 mg/dL); IgM, 206 mg/dL (normal range, 35 to 220 mg/dL); IgA, 208 mg/dL (normal range, 110 to 410 mg/dL); rheumatoid factor, 51 IU/L (normal range, <15.0 IU/L); CH50, 17 U/mL (normal range, 30 to 50 U/mL); C3, 53 mg/dL (normal range, 68 to 128 mg/dL); and C4, 16 mg/dL (normal range, 14 to 36 mg/dL). No immune complex (IC) was detected by the C1q-binding assay. ANA titer was ×1,280 with a speckled pattern and positive results were obtained for anti-SSA antibody and anti-RNP antibody. Negative results were obtained for all other autoimmune antibodies, including anti-SSB, anti-dsDNA, anti-Sm, anti-cardiolipin, anti-centromere, anti-topoisomerase, anti-glomerular basement membrane, and anti-mitochondrial antibodies. Negative results were also obtained for myeloperoxidase and proteinase-3 antineutrophil cytoplasmic antibodies. Urinalysis revealed proteinuria and hematuria. Urinary protein excretion rate was 4.21 g/day. Arterial blood gas analysis in room air showed: pH, 7.497; pO2, 72.8 Torr; pCO2, 28.4 Torr; and HCO3-, 21.8 mmol/L. A chest radiograph showed bilateral, slightly reticular opacities predominantly in the middle and lower lung fields (Fig. 2A). A high-resolution chest computed tomography (CT) showed bilateral ground-glass opacities, with relative sparing of the peripheral lung parenchyma (Fig. 2B). Pleural effusion and ascites were absent on the thoracoabdominal CT. Diffuse alveolar hemorrhage (DAH) was diagnosed, based on hemoptysis, chest CT findings and the rapid decrease in Hb from 10.9 to 8.3 g/dL in 3 months. Because both the hemoptysis and dyspnea gradually worsened, the patient was treated with methylprednisolone (methyl PSL; 1 g/day for 3 days) and subsequently PSL (60 mg/day for 4 weeks). Afterward, the PSL dosage was gradually reduced every 2 weeks. The respiratory symptoms and hypoxia ameliorated rapidly within 1 week. Approximately 4 weeks after starting methyl PSL treatment, a renal biopsy was performed. Light microscopy of the renal biopsy specimens (stained with hematoxylin and eosin) showed no significant glomerular findings (Fig. 3A), although, periodic acid methenamine silver staining showed spike formation in the basement membrane (Fig. 3B). In addition, immunofluorescence microscopy showed diffuse granular deposits of IgG (Fig. 3C) and C3 (Fig. 3D) in the basement membrane. The results of the renal biopsy examinations were compatible with lupus nephritis (World Health Organization, class V). With the findings from the renal biopsy examinations and the positive results for ANA, SLE was diagnosed, based on the Systemic Lupus International Collaborating Clinics classification criteria for SLE (the Systemic Lupus International Collaborating Clinics [SLICC] classification criteria) [2], rather than the revised ACR criteria. DAH as well as PLE and vitiligo were considered to be due to SLE. At this point, secondary Sjögren syndrome was also diagnosed because of sicca symptoms and positive results in Schirmer test and sialography. However, no other collagen disease, including mixed connective tissue disease or vasculitis could be diagnosed because of the lack of positive criteria and the positive findings in the renal biopsy examinations. Because the proteinuria and serum hypoalbuminemia had not fully ameliorated, cyclosporin A (100 mg/day) was added to PSL (40 mg/day) 7 weeks after initiation of steroid therapy. Approximately 4 months after starting steroid therapy, the above-mentioned symptoms and laboratory data, including pO2, serum Alb, Hb and urinalysis almost normalized. A chest radiograph and a high-resolution chest CT showed no abnormal findings (Fig. 2C, D).
Discussion
SLE is a chronic autoimmune disease predominantly occurring in women under the age of 50 years involving multiple organ systems, particularly kidneys, cardiovascular system, skin, and central nervous system. Age at onset has been recognized as having a modifying effect on the clinical manifestations of SLE, with late onset in individuals aged ≥50 years possibly constituting a specific subgroup of SLE [3]. The age-related variability in disease expression remains unclear although the altered responsiveness of the aging immune system may be involved. Moreover, it has been speculated that older and younger patients may have different genetic determinants of disease and respond to different triggering mechanisms. Alternatively, the less-active expression of SLE both clinically and immunologically in older patients may reflect senescence of the immune system [4].
In late-onset SLE patients, because the onset is insidious, the interval between onset and diagnosis is longer than in younger patients [3]. With regard to clinical features in late-onset SLE, serositis, lung involvement and Sjögren syndrome are more prevalent, whereas skin manifestations, photosensitivity, arthritis and nephritis are uncommon [5].
Prior studies have suggested that late-onset SLE is associated with a benign clinical course and has a better prognosis than SLE in younger patients [5,6]; however, some authors have considered that the degree of organ damage is greater in late-onset SLE [7,8]. Tomic-Lucic et al. [9] reported that late-onset SLE had poorer prognosis because of the high frequency of comorbid conditions and more severe organ damage resulting from aging and longer exposure to classical vascular risk factors. Lalani et al. [10] indicated that disease activity and damage accrual were higher in the adult-onset SLE patients, despite less lupus nephritis. They also suggested that adult-onset SLE was not benign. Mak et al. [8] also reported the clinical profile of late-onset SLE did not constitute a benign subgroup of the lupus population. With regard to laboratory findings, late-onset SLE shows lower rates of immunological disorder, including anti- dsDNA antibody, anti-RNP antibody, and hypocomplementemia [11]. A period of 9 years was required to diagnose SLE in the case presented in this report. In regard to the distinctive findings in the present case, PLE, vitiligo, and DAH, which are all rarely seen in SLE, not only occurred but were also present concurrently at various points. In particular, the coexistence of SLE and vitiligo has very rarely been reported. In these previously reported cases, two of three patients were diagnosed with SLE in their twenties and showed complications of vitiligo as well as cardiomyopathy and renal failure [12]. The other patient was a 64-year-old woman with complications of vitiligo and pernicious anemia [13]. Nath et al. [14] identified chromosome 17p13 as the genomic region that appears to contribute to SLE in members of families who inherit lupus together with vitiligo. Because the patient’s father had also suffered from late-onset SLE, genetic factors involving this chromosomal region might have been associated with the emergence of SLE. The pleural effusion and ascites due to PLE did not worsen and moreover, they resolved without steroid therapy on this visit, and the patient also recovered from DAH, a life-threatening condition [15]. Considering these clinical findings and the negative laboratory findings for anti-dsDNA antibody, disease activity in the present case might have been weak, potentially contributing to the delay in diagnosis. On this visit, 9 years after initial symptoms occurred, SLE was finally able to be diagnosed. In the present case, SLE was diagnosed on the basis of the SLICC classification criteria, not the revised ACR criteria. With the use of the SLICC classification criteria, SLE can be diagnosed on the basis of biopsy-confirmed nephritis compatible with SLE and positive results for ANA. The SLICC classification criteria show better performance than the revised ACR criteria in terms of sensitivity, but not specificity. These criteria are meant to be more clinically relevant, allowing the inclusion of more patients with clinically-defined lupus than the current ACR criteria. Because late-onset SLE has a longer lag time to diagnosis because of the atypical physical and laboratory features, the SLICC classification criteria may be helpful for the earlier diagnosis of late-onset SLE.