서 론
대장암은 전 세계적으로 세 번째로 흔하게 발생하는 악성 종양으로 암으로 인한 사망의 주요 원인으로 알려져 있다[1]. 대부분의 대장암은 산발성 대장암(sporadic colorectal cancer)으로 유전적 요인이 관여하지 않지만, 전체 대장암의 약 15%–30%는 유전적 요인에 의해 대장암이 발생하게 된다. 이 중 약 10%–25%는 원인 유전자가 명확하지 않지만 가족력 등 유전적 경향을 보이는 가족성 대장암에 해당하고, 나머지 2%–5%는 원인 유전자가 알려져 있는 유전성 대장암에 해당한다[2]. 유전성 대장암은 크게 유전성 비용종증 대장암(hereditary nonpolyposis colorectal cancer; HNPCC)과 유전성 용종 증후군(hereditary polyposis syndrome)으로 나눌 수 있고, 유전성 비용종증 대장암이 대부분을 차지하고 있다.
유전성 비용종증 대장암은 상염색체 우성으로 유전되는 질환으로 1913년 Aldred Warthin에의해 처음으로 보고되었다[3]. 전체 대장암의 약 5%에 해당하며[4], 해당 환자들은 대장암의 평생 이환율이 약 80%에 이르는 것으로 보고되고 있다[5]. 유전성 비용종증 대장암 환자들은 일반적인 산발성 대장암 환자들에 비해 조기에 대장암이 발생하고, 대장암 외에 비뇨생식기나 위장관 등에 다른 악성 종양이 흔하게 동반된다[5]. 유전성 비용종증 대장암은 린치증후군(Lynch syndrome)으로도 알려져 있으며, 대장암만 특이적으로 발생하는 린치증후군 I과 대장암과 타장기암이 함께 발생하는 린치증후군 II로 구분하기도 한다[6]. 유전성 비용종증 대장암은 유전성 용종 증후군과 달리 심한 대장 용종증을 잘 동반하지 않아 병변만으로는 산발성 대장암과의 구별이 어려우나 그 발생 원인이나 임상적 특징, 치료나 예후 측면에 있어 서로 다른 특징을 보이게 되므로 유전성 비용종증 대장암에 대한 정확한 이해와 적절한 접근이 필요할 것으로 생각된다. 이에 본 종설에서는 유전성 비용종증 대장암의 임상적 특징과 발생 원인, 진단과 치료 등 유전성 비용종증 대장암의 전반적인 내용에 대해 기술하고자 한다.
본 론
유전성 비용종증 대장암은 유전성 용종 증후군과 달리 심한 대장 용종증을 잘 동반하지 않으며 대장암의 평생 이환율은 약 80% 정도로 보고되고 있다[5,7]. 산발성 대장암에 비해 조기에 대장암이 발생하여 50세 이전에 대장암을 진단받는 경우가 흔하다[8]. 유전성 비용종증 대장암도 산발성 대장암과 마찬가지로 전구 병변인 선종으로부터 대장암이 발생하게 되는데, 산발성 대장암의 선종에 비해 크기가 크고, 융모성 선종의 빈도가 높으며, 고도의 이형성증을 동반하는 경우가 많다[9]. 또한 산발성 대장암의 경우 선종으로부터 암으로의 진행 속도가 10–15년인데 비해, 유전성 비용종증 대장암의 경우는 그 기간이 약 3년 정도로 빠르다[9,10]. 또한 조직학적 분화도가 나쁘고 점액암이 자주 관찰되는 등의 소견을 보이나 산발성 대장암에 비해 예후는 좋은 것으로 알려져 있다[11]. 산발성 대장암에 비해 우측 대장에서 호발하고, 동시성 대장암과 이시성 대장암의 발생 위험이 더 높다. 주로 2기에서 진단되는 경우가 많고 현미부수체 불안정성(microsatellite instability)이 흔하다[11].
유전성 비용종증 대장암 환자에서는 대장암 외에도 여러 타장기암의 발생 위험도가 높아지는데 이 중 자궁내막암이 가장 흔하며 여자의 경우 대장암과 비슷한 발생 위험도를 보이게 된다[7,11]. 이 외에도 위암, 난소암, 췌담도암, 요관암, 소장암, 유방암, 전립선암 등이 동반되기도 한다[7,11]. 이처럼 유전성 비용종증 대장암은 대장암을 비롯한 여러 암이 함께 발견되는 증후군이며, 해당 환자의 가족에서 비슷한 종류의 암이 자주 발견되는 특징을 보이게 된다.
유전성 비용종증 대장암은 상염색체 우성으로 유전되는 질환으로 복제실수교정 유전자(mismatch repair gene)의 배선돌연변이(germline mutation)에 의해 발생한다[12,13]. 복제실수교정 시스템은 DNA 복제와 재조합 과정에서 발생할 수 있는 염기의 복제실수를 인지하고 교정하는 시스템으로, 이 시스템에 결함이 있는 경우에는 현미부수체 불안정이 발생하고 이는 발암과정의 주요 기전의 하나로 알려져 있다[14,15]. 이러한 복제실수교정 시스템을 조절하는 대표적인 유전자로 MLH1, MSH2, MSH6, PMS2가 있으며, 이 중 MLH1과 MSH2의 변이가 유전성 비용종증 대장암 환자의 약 90%에서 발견된다. 약 10%의 환자에서 MSH6의 변이가 발견되며, PMS2의 변이는 매우 드물게 발생한다[16,17]. 변이 유전자에 따른 임상 양상의 차이는 아직까지 잘 알려져 있지는 않으나 일부 연구들에 따르면 MSH6에 변이가 있는 경우에 다른 유전형에 비해 대장암의 발생위험은 낮은 반면 자긍내막암의 발생 위험은 상대적으로 더 높은 것으로 보고되기도 하였다[18].
최근 연구에 따르면 EpCAM 유전자에 배선돌연변이가 있는 경우도 유전성 비용종증 대장암이 발생할 수 있는 것으로 보고되었다. EpCAM은 MSH2의 앞부분(upstream)에 위치하고 있는데, EpCAM의 3’ 영역에 배선유전자 결실(germline deletion)이 발생하면 EpcAM-MSH2 유전자 융합전사물(fusion transcript)이 생기고, 이로 인해 복제실수교정 유전자 자체에 변이는 없지만 MSH2의 프로모터 부위가 과메틸화(hypermethylation) 되어 MSH2의 발현이 억제됨으로써 유전성 비용종증 대장암이 발생하게 되는 것이다[19-21]. 이러한 EpCAM의 변이에 의한 경우는 전체 유전성 비용종증 대장암의 6.3%에 해당하는 것으로 보고되고 있다[22].
유전성 비용종증 대장암을 진단하는데 있어서는 가족력 조사, 현미부수체 불안정성 검사, 면역조직화학 검사를 통한 복제실수교정 유전자의 발현 평가, 유전자 검사 등 여러 가지 방법이 이용되고 있다[11]. 이 중 가계도 분석을 통한 가족력 조사는 유전성 비용종증 대장암을 진단하는데 있어 가장 기본적이고 중요한 방법이다. 특히 일반인에 비해 대장암이 일찍 발생하거나 타장기 암이 동반되어 있거나 가족 중에 비슷한 암이 자주 발생한 경우에는 유전성 비용종증 대장암을 의심해야 한다. 이와 관련해 대장암 국제협력기구에서는 유전성 비용종증 대장암을 진단하기 위한 임상지침을 마련하기 위해 암스테르담 진단기준 I을 발표하였으나 실제로 유전성 비용종증 대장암 환자의 50% 이상에서 이 기준을 만족하지 못하는 문제점이 제기되었다. 따라서 현재는 진단 기준을 좀 더 완화한 암스테르담 진단기준 II와 수정된 베데스다 진단기준을 주로 사용하고 있다(Table 1) [23,24]. 하지만 이는 유전성 비용종증 대장암을 확진하기 위한 것이 아니라 추가검사가 필요한 의심 환자군을 선별하기 위한 기준으로 사용되는 것임을 알아야 한다.
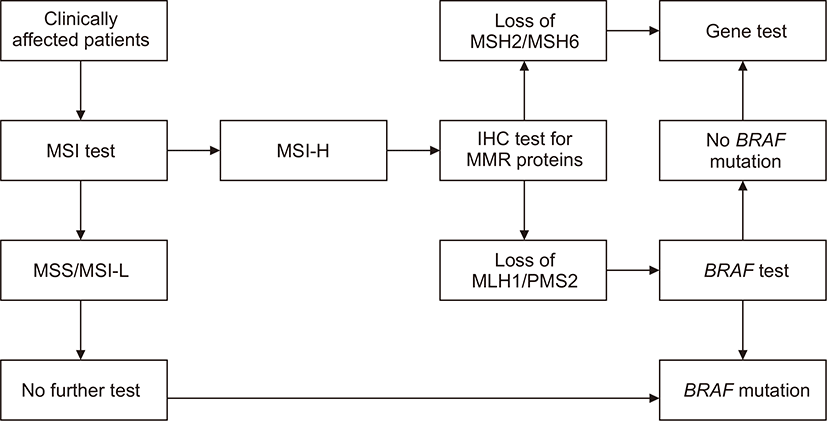
이러한 임상 진단기준에서 유전성 비용종증 대장암이 의심되는 경우 현미부수체 불안정성 검사를 시행할 수 있다. 현미부수체 불안정성은 복제실수교정 유전자의 변이로 인한 과다돌연변이성 상태를 일컫는 말로 현미부수체의 반복서열이 비정상적으로 확장되거나 단축되는 상태를 말한다[25,26]. 현미부수체 불안정성 검사에는 2개의 단염기 현미부수체인 BAT23, BAT26과 3개의 쌍염기 현미부수체인 D5S346, D2S123, D17S250로 구성된 베데스다 패널이 주로 사용되어 왔다. 최근에는 단염기 현미부수체인 BAT25, BAT26, NR21, NR24, NR22 (또는 NR27)로 구성된 새로운 패널을 제시하여 기존의 베데스다 패널보다 민감도와 특이도를 높였다[27,28]. 이 패널 중 두 개 이상의 현미부수체가 불안정성을 보이는 경우를 MSI-H (high)로 정의하며, 하나의 현미부수체에서만 불안정성을 보이는 경우를 MSI-L (low), 불안정성을 보이는 현미부수체가 하나도 없는 경우를 MSS (stable)라고 한다[29,30]. MSI-H인 경우를 현미부수체 불안정성이 있다고 평가하며 산발성 대장암의 경우 10-15%가 MSI-H인 반면, 유전성 비용종증 대장암의 경우에는 거의 대부분의 환자가 MSI-H이기 때문에 현미부수체 불안정성 검사는 추가적인 유전자 검사가 필요한 대상군을 선별하는 데 사용될 수 있다.
유전성 비용종증 대장암이 의심되는 경우 유전자 검사 대상을 선별하기 위한 또 다른 방법으로 복제실수교정 단백질에 대한 면역조직화학 검사를 들 수 있다. 이는 대장암 조직에 대해 MLH1, MSH2, MSH6, PMS2 단백질의 발현 여부를 평가하기 위한 것으로 면역화학염색 상 특정 단백질이 염색이 되지 않는 경우에 해당 유전자에 변이가 있음을 시사한다[31,32]. 하지만 산발성 대장암의 경우에도 MLH1 단백질의 발현이 소실되는 경우가 있으므로 이에 대한 확인이 필요하다. 산발성 대장암에서는 유전성 비용종증 대장암의 경우와 달리 BRAF 유전자의 V600E에 체세포 돌연변이로 인해 MLH1 유전자의 프로모터가 과메틸화됨으로써 MLH1 단백질의 발현이 소실되게 된다[9,33]. 따라서 면역조직화학 검사에서 MLH1 단백질의 발현이 소실된 경우에는 BRAF 검사를 시행하여 산발성 대장암의 가능성을 확인해야 한다.
유전성 비용종증 대장암을 확진하기 위한 최종 검사는 유전자 검사다. 유전자 검사는 유전성 비용종증 대장암의 원인이 되는 복제실수교정 유전자인 MLH1, MSH2, MSH6, PMS2의 배선돌연변이 여부를 확인하는 것으로 염기서열분석 방법이 주로 사용된다[16,34]. 복제실수교정 유전자 배선돌연변이의 90% 이상이 MLH1과 MSH2에서 발견되므로 이 두 유전자에 대한 검사를 우선 시행하는 것이 효과적일 수 있다. 또한 유전자 검사에서 복제실수교정 유전자의 변이가 발견되지 않은 경우에는 EpCAM 유전자의 결실이 있는지 확인해야 한다. 유전자 검사는 유전성 비용종증 대장암을 확진할 수 있는 유용한 검사이지만, 시간과 비용이 많이 소요되므로 앞서 기술한 선별검사를 통해 유전자 검사가 필요한 대상군을 적절히 선정하는 과정이 필요하다(Fig. 1).
유전성 비용종증 대장암 환자를 진단하는 것이 중요한 이유는 이 질환에 이환된 환자와 환자의 가족에 대해 대장암을 비롯한 타장기암에 대한 고위험군을 찾아내고, 정기검진을 통해 암의 발생을 조기에 진단하고 치료하기 위함이다. 복제실수교정 유전자의 배선돌연변이가 발견된 가족 구성원 및 해당 환자에 대해서는 대장내시경, 산부인과 검진, 위내시경, 초음파검사 등 유전성 비용종증 대장암에서 호발하는 각종 암에 대한 정기 검진이 필요하다. 대장내시경의 경우 20–25세에 첫 검진을 시작하여 1-2년 간격으로 정기 검진을 받도록 권고하는 등 일반인과 다른 진료지침을 권고하고 있다(Table 2) [9,34,35].
유전성 비용종증 대장암의 경우 산발성 대장암과 같이 대장의 부분절제를 시행할 경우에 수술 후 약 50%의 환자에서 이시성 대장암이 발생하는 것으로 보고되고 있어 최소한 아전결장절제술을 시행해야 한다[36,37]. 이는 이시성 대장암의 발생 위험을 줄이면서 동시에 직장을 보존함으로써 배변기능을 유지할 수 있는 장점이 있다[11]. 하지만 이들에서 직장암의 발생 위험도가 10%–15% 정도로 보고되고 있으므로 수술 후 주기적인 직장경 검사가 반드시 필요하다[38]. 유전성 비용종증 대장암으로 인한 직장암의 경우에는 전직장 절제를 포함한 전대장절제술 및 회장항문문합술을 시행해야 한다[9,39]. 또한 국소진행성 직장암인 경우에는 수술 후 회장낭 기능의 보존을 위해 수술 후 항암방사선 치료를 하기보다는 수술 전 항암방사선 치료를 하는 것이 바람직하다[39]. 그리고 대장암이 발견되지는 않았지만 복제실수교정 유전자의 배선돌연변이가 있는 환자의 경우, 예방적 대장 절제술은 아직까지는 시행되고 있지는 않다[14].
유전성 비용종증 대장암에서 타장기암의 경우에는 산발성 암의 치료원칙과 크게 다르지 않으나, 여성의 경우 자궁내막암과 난소암의 위험이 높으므로 가임기가 아니거나 임신의 계획이 없다면 대장암 수술 시 예방적으로 자궁적출술 및 양측 난소절제술을 함께 시행하는 것이 바람직하다[40-42].
유전성 비용종증 대장암은 산발성 대장암에 비해 예후가 더 좋은 것으로 알려져 있으며, 수술 후 보조항암치료의 원칙은 산발성 대장암과 다르지 않다. 다만 MSI-H인 경우 5-fluorouracil에 대한 반응성이 낮은 걸로 보고되고 있지만 이에 대해서는 여전히 논란이 있다[14,43].
결 론
유전성 비용종증 대장암은 원인유전자가 밝혀진 유전성 대장암 증후군 중 가장 흔한 유형으로 이들에 대한 정확한 이해와 적절한 관리가 필요하다. 유전성 비용종증 대장암이 임상적으로 의심되는 경우 유전자 검사를 통해 정확히 진단해야 하고, 유전상담 등을 통해 환자 개인뿐 아니라 해당 가족 구성원에 대한 동시적 접근이 필요하다. 유전성 비용종증 대장암에 이환된 환자 및 가족 구성원은 권고안에 따른 정기 검진을 시행함으로써 관련 암의 발생을 조기에 진단하고 치료해야 한다. 유전성 비용종증 대장암은 산발성 대장암에 비해 예후가 좋으므로 적절한 예방과 관리를 통해 생존율 향상에 기여할 수 있을 것으로 기대된다.