Introduction
Idiopathic renal hypouricemia (RHUC) is a rare hereditary disease caused by impaired uric acid transporter, reabsorption insufficiency and/or secretion acceleration. There are two types of RHUC: type 1 (RHUC1) [1,2], which is caused by a mutation in the SLC22A12 gene that encodes a renal urate-anion exchanger, URAT1; whereas type 2 (RHUC2) was previously found to be caused by a defect in the SLC2A9 gene, which encodes a high-capacity glucose and urate transporter, named glucose transporter (GLUT9) [3]. Whereas most patients are clinically asymptomatic and are detected incidentally, exercise-induced acute kidney injury (EIAKI), urolithiasis, or hematuria may develop in some patients [4-6]. The present study describes the case of a patient with familial RHUC who presented with severe acute kidney injury (AKI) and had a homozygous pathogenic variant of SLC22A12 gene.
Case
The patient provided written informed consent for the publication and the use of his images.
A 17-year-old male was transferred to our hospital because of azotemia, decreased urine output, and abdominal pain for 2 days. His past history and family history were unremarkable including no vigorous exercise habit and acute kidney injury except high protein diet for 6 months.
At admission, his body weight was 83.8 kg (90th to 95th percentile), height 169.2 cm (25th to 50th percentile) and body mass index 29.2 kg/m2 (>97th percentile). His blood pressure was 171/100 mmHg, pulse rate was 101 beats/min, respiratory rate was 22/min, and body temperature was 37.1℃. Laboratory data at admission showed the following results: hemoglobin 14.3 g/dL, WBC count 9,750/μL, platelet 253,000/μL, sodium 141 mEq/L, potassium 4.0 mEq/L, total CO2 18 mEq/L, BUN 35 mg/dL, creatinine (Cr) 6.13 mg/dL, uric acid 4.9 mg/dL, calcium 8.6 mg/dL, phosphorus 4.5 mg/dL, AST 16 IU/L and ALT 18 IU/L. The urinalysis revealed a specific gravity of 1.008, a pH of 5.5, protein of 2+, blood of 1+, RBC 11-20/HPF. The 24-hour urinary protein was 13.25 mg/m2/hr, Cr clearance was 31.3 mL/min/1.73m2 and fractional excretion of uric acid was increased to 70.27%. The following parameters were within normal limits or negative: serum immunoglobulin; serum complements; antinuclear antibody and anti-neutrophil cytoplasmic antibodies.
Abdominal ultrasonography showed increased echogenicity of both renal parenchyma and poor cortical medullary differentiation. The initial impression was rapidly progressive glomerulonephritis. Ultrasonography guided kidney biopsy followed by methylprednisolone pulse therapy were conducted. However, Cr elevated to 9.26 mg/dL on 3rd hospital day. Hemodialysis was applied for 2 days until renal function recovered.
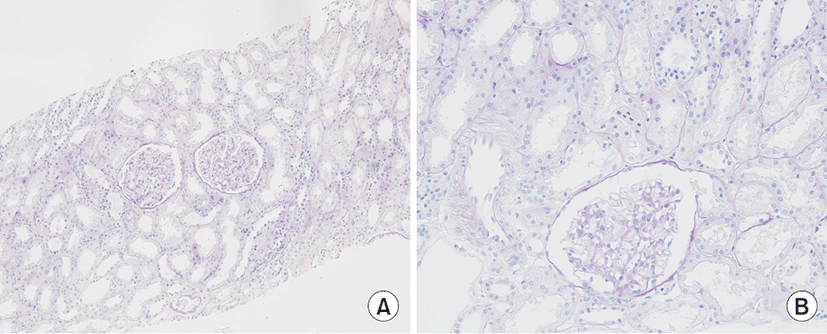
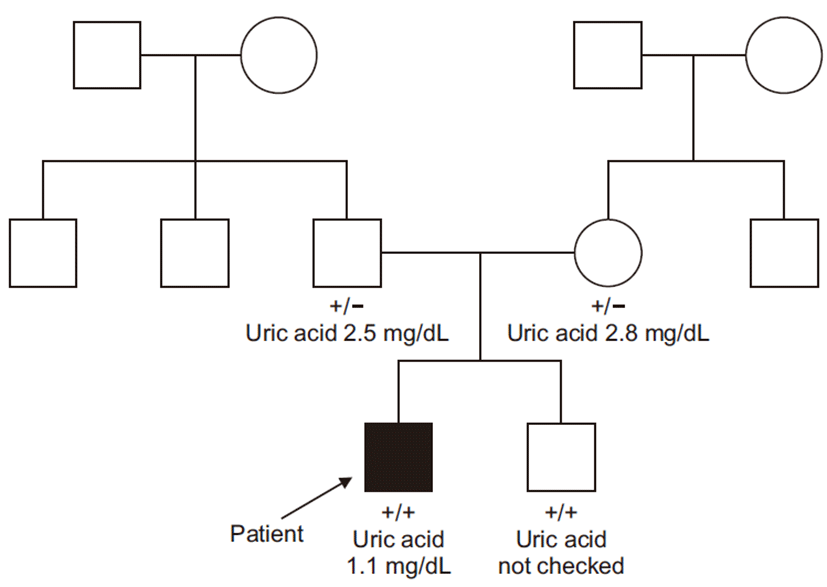
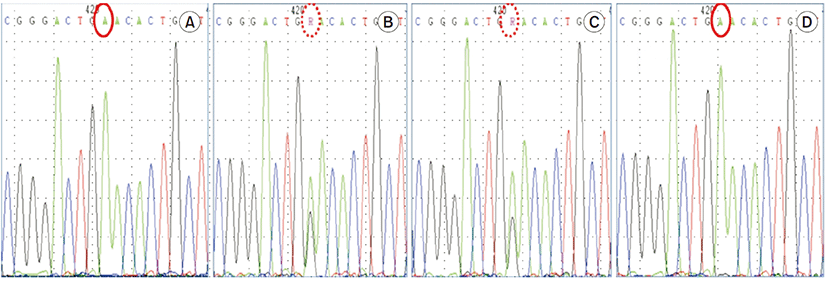
Histopathological findings revealed normal glomeruli and arterioles without crescent and tubulointerstitial abnormalities. Immunofluorescence showed were all negative (Fig. 1). Due to normal finding in renal biopsy and continuously low serum uric acid as low as 1.0 mg/dL, genetic study, sequence analysis of the SLC22A12 gene, was performed in all family members (Fig. 2). The patient carries a homozygous pathogenic variant of SLC22A12 gene such as NM_44585.4(SLC22A12):c.774G>A, p.Trp258Ter. Both parents are heterozygous for the pathogenic variant, while his brother carries same homozygous pathogenic variant (Fig. 3). During the outpatient follow up, serum uric acid was low (0.7–1.1 mg/dL) and renal function was normal (Table 1).
Discussion
RHUC is defined as a lower serum uric acid concentration caused by decreased production or increased excretion, and idiopathic RHUC is a familial hereditary disease characterized by an increased renal urate clearance cause by an isolated inborn error of membrane transport for urate in the proximal renal tubules [1,2].
The diagnosis of RHUC is based on biochemical markers: hypouricemia (<2.0 mg/dL) and increased fractional excretion of uric acid (>10%) without evidence of secondary causes of hyperuricosuric hypouricemia (such as Wilson disease, Fanconi syndrome, and drug-induced tubulopathy) [7]. The diagnosis can be confirmed by molecular analysis of the mutations in the SCL22A12 and/or SCL2A9 genes.
Ever since Akaoka et al. [8] first reported a case study of RHUC in Japan in 1975, RHUC has been known to be relatively common in Japanese population (approximately 0.3%). The incidence of RHUC has been reported to 0.12% to 0.72%.
Enomoto et al. [1] identified a uric acid transporter in the human kidney, URAT1, encoded by SLC22A12. URAT1 is a uric acid anion exchanger which regulates blood uric acid levels and which is targeted by uricosuric and antiuricosuric agents. Most patients with idiopathic RHUC have loss of function mutations in SLC22A12. About half of those with SLC22A12 mutations were homozygotes, about a third were compound heterozygotes, and the rest were heterozygotes [9]. In Japanese and Koreans, the W258X mutation is reported as the predominant genetic cause of RHUC [9,10].
In Korean case reports, Cheong et al. [9] reported five idiopathic renal hypouricemia patients and their families; two were hematuria, one was ureter stone, one was EIAKI, and one was incidentally detected. SLC22A12 gene mutations (W258X, R90H, and R477H) were found in four families, but not in one family. Kim et al. [11] reported familial renal hypouricemia confirmed by genotyping of SLC22A12 with EIAKI in 24-year-old male. Although most patients with RHUC have no clinical symptoms or complications, the major complications are EIAKI, urolithiasis and hematuria [11].
Potential mechanisms of AKI include increased uric acid production during exercise causing urate nephropathy, renal vasoconstriction following depletion of free radical scavengers including urate, or increased free radicals with ischemia reperfusion injury. During episodes of AKI, serum uric acid levels are inappropriately normal or low, as in this case [12].
In this study, possible explanation of the severe AKI could be due to habitual consumption of excess dietary protein provoking acute renal disease through increased glomerular pressure/hyperfiltration and renal hypertrophy.
Our case showed some difference from previous cases. First, the patient showed features of rapidly elevated serum Cr and low serum uric acid without vigorous exercise history nor familial AKI history. Second, the rise of serum Cr to 9.26 mg/dL on the third day of hospitalization was normalized after hemodialysis. Finally, the renal histopathologic examination showed normal findings despite severe AKI.
Based on this, genotyping was conducted to determine the RHUC, revealing a homozygous pathogenic variant such as NM_144585.4(SLC22A12):c.774G>A, p.Trp258Ter. Both patents are heterozygous for the mutation of W258X, while his brother carries same homozygous mutation. Compared to the patient, both parents showed a normal level of uric acid and a normal renal function. The clinical features of AKI with unknown cause in this patient, and the genotyping result, confirmed the diagnosis of familial RHUC.
In conclusion, familial RHUC is a rare disorder but must be considered in a patient with a low serum uric acid levels and AKI, family history of urolithiasis and EIAKI. SLC22A12 mutation screening is an important molecular investigation for the diagnosis of familial RHUC, and should be conducted in patients suspected to have the disease.