Introduction
Pseudohypoaldosteronism (PHA) comprises of a heterogeneous group of disorders of electrolyte metabolism characterized by an apparent state of renal tubular unresponsiveness or resistance to the action of aldosterone. It is manifested by presence of salt wasting, hyperkalemia, and metabolic acidosis [1]. Signs and symptoms of PHA can be rather unspecific and include poor feeding, poor weight gain or failure to thrive, vomiting, diarrhea, polyuria, and dehydration [2]. PHA is classified as primary and secondary type. Secondary transient PHA has been reported in infants with urinary tract infection (UTI) and urinary tract malformation. The acute biochemical presentation of PHA is identical to adrenal crisis due to congenital adrenal hyperplasia (CAH) and at initial stages the condition is suspected as CAH.
Here, we report an infant who presented with failure to thrive and finally diagnosed with transient PHA due to UTI with vesicoureteral reflux.
Case
A 5-month-old female infant was initially transferred to our hospital for evaluation of poor weight gain for 2 months. The body weight of the infant was 5.0 kg (<3th percentile) and the height was 57.3 cm (<3th percentile). Her birth weight was 2.97 kg (10th to 25th percentile) and gestational age was 39 weeks with normal spontaneous vaginal delivery. The infant had been breastfeeding and weighed 5 kg at 3 months of age but developed no weight gain after that.
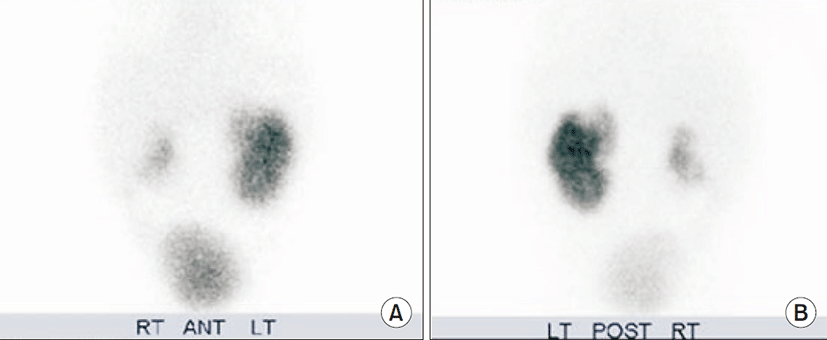
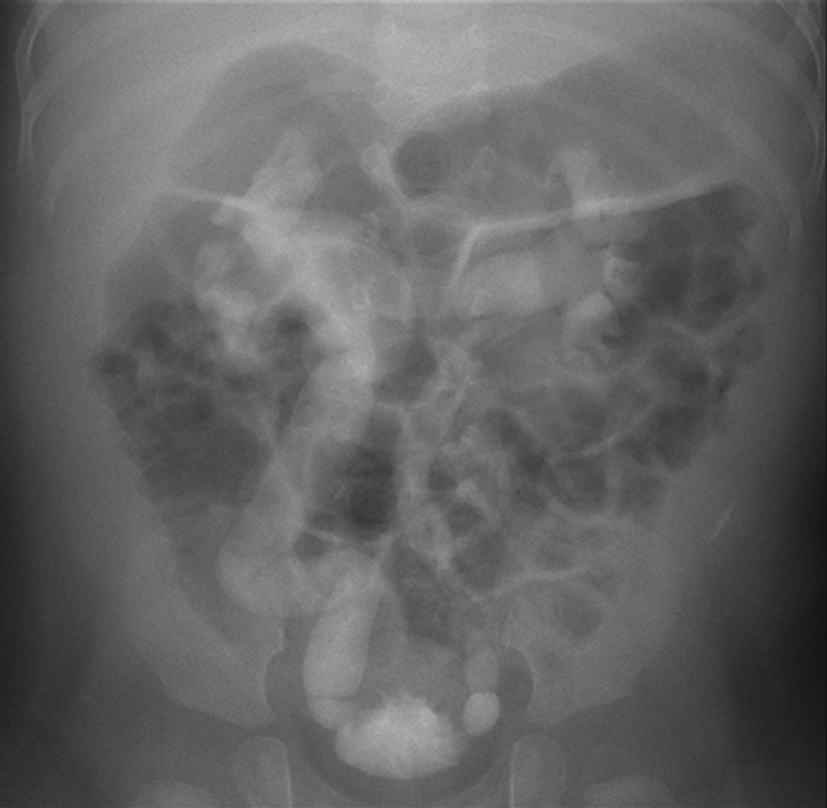
On physical examination, she was afebrile and had not so ill looking appearance. Body temperature was 36.9oC, pulse rate 120 beats/min. respiration rate 40 beats/min, and blood pressure was 85/60 mmHg. Virilization of external genitalia or pigmentation were not remarkable. Initial serum sodium was 125 mEq/L and serum potassium was 6.1 mEq/L. The serum CRP level was 4.08 mg/dL and serum ESR level was 60 mm/hr. Urine analysis revealed pyuria. Intravenous saline and antibiotics were started after examination of urine culture. Catheterized urine culture was positive for Serratia marcescens. The initial serum 17-hydroxyprogesterone level was 0.73 ng/mL (normal range, 0.13 to 0.106 ng/mL) and aldosterone level was markedly elevated (17,800 pg/mL) (normal range, 50 to 900 pg/mL), and urinary sodium was 30 mg/L. The changes in clinical parameters led to apparent diagnosis of PHA. The infant’s serum sodium and potassium levels were normalized after 48 hours of intravenous sodium replacement and antibiotics therapy, and inflammatory markers were normalized. The renal ultrasonography showed mild atrophy of right kidney and compensatory hypertrophy of left kidney. Technetium-99m-dimercaptosuccinic acid renal scan demonstrated multiple renal cortical defects in both kidneys and generalized areas of diminished uptake in right kidney (Fig. 1). The voiding cystourethrography showed right-sided grade V and left-sided grade IV vesicoureteral reflux (Fig. 2). The follow-up electrolyte levels remained within normal range and aldosterone level was decreased without oral sodium replacement and proper weight gain was achieved during outpatient follow-up.
Discussion
A variety of factors contribute to the pathogenesis of hyponatremia and hyperkalemia in neonatal and infant periods. In these children is very important to establish or exclude CAH because this is a life-threatening condition [3]. More than 90% of CAH are caused by 21-hydroxylase deficiency (so-called classical salt wasting form). The clinical presentation of 21-hydroxylase deficiency in female results in masculinized external genitalia and vomiting within a few days/weeks after birth, severe renal wasting, and dehydration in both sexes. Our patient was an infant with poor weight gain and with normal external genitalia. He showed hyponatremia, hyperkalemia and elevated serum aldosterone level.
PHA is subdivided into primary (genetic) and secondary (or transient) forms. Primary PHA has two clinically and genetically distinct forms with either an autosomal recessive or an autosomal dominant disorder [4]. The autosomal recessive form (the so-called multiple organ form) is associated with mutation in the subunit of the epithelial sodium channel (ENaC gene), causing salt wasting not only from the kidney, but also from the colon, sweat glands, and salivary glands [1,5]. The autosomal dominant form is due to mutation in the mineralocorticoid receptor gene, which prevents normal receptor function and, hence, causes renal salt wasting [6].
A secondary form of PHA, limited to the kidney, may be associated either with medications or tubulointerstitial disease. Secondary PHA has been described most frequently in neonates and young infants in association with UTI and/or urinary tract malformations such as, hydronephrosis, ureteropelvic junction obstruction, vesicoureteral reflux, or posterior urethral valve [7,8]. After treatment for UTI and/or surgical therapy, disappearance of secondary aldosterone resistance has been noted.
Rodriguez-Soriano et al. [7] first described the entity of transient PHA to be associated with obstructive uropathy in 1983. The mechanism is poorly understood, but it is speculated that it may be due to parenchymal scarring secondary to obstruction and tubular aldosterone resistance secondary to endotoxin damage of the aldosterone receptors from cytokine such as, TGF-β [9]. Early infancy, obstructive uropathy, and UTI are three major risk factors for the development of secondary PHA. Most of the patients are younger than 3 months of age, which suggests that immature renal tubules may be responsible for the development of resistance to aldosterone. Furthermore, the severity of salt wasting is inversely correlated with age, thus supporting a role for tubular immaturity [10]. Bogdanovic et al. [2] reported that 90% of patients with transient PHA were less than 3 months of age and 89% of them suffered from urinary tract malformation associated with UTI. Melzi et al. [11] never observed transient PHA in infants older than 3 months despite the presence of urinary tract malformation.
Bowden et al. [12] first found that autosomal dominant PHA with a mineralocorticoid receptor gene (NR3C2) mutation, presenting with salt wasting crisis was associated with UTI and posterior urethral valves, thereby mimicking secondary PHA [13]. If serum aldosterone is persistently elevated after UTI treatment, then NR3C2 gene testing should be considered to identify the cause, to determine future recurrence risk, and to prevent serious salt wasting in a subsequent family member. It should be noted that our patient was 5-month-old infant and only manifested poor weight gain and failure to thrive since 3 months of age. The differentiation between CAH and PHA is difficult because the electrolyte imbalances are identical, however virilization or pigmentation is not found in PHA. Delayed antibiotics treatment in patients with UTI misdiagnosed as CAH could be fatal because of the life-threatening risk of septic shock. Therefore, it is mandatory to include urinalysis and abdomen ultrasound in work-up of patients with salt wasting to exclude infection or urinary tract malformation [14].
Transient PHA should be suspected in an infant with severe hyponatremia, hyperkalemia and failure to thrive without virilization or pigmentation. Urinalysis and urinary tract imaging studies should always be performed in order to allow early diagnosis of transient PHA, thus preventing serious complications and inappropriate treatments.