
Introduction
Dementia is a progressive neurodegenerative disease that not only profoundly impairs the health and well-being of patients, but also poses a major public health problem. Alzheimer disease (AD) is a common neurodegenerative disorder characterized by a gradual deterioration in cognitive performance, particularly in the memory domain, which impacts the daily functioning and life of the individual. The prevalence of AD increases with age, doubling every 5 years from the age of 60 years [1]. This disease is marked by compromised cognitive function, often associated with a significant reduction in brain volume [2]. Another characteristic of the disease is the presence of amyloid beta (Aβ) plaques in the brain, which result from the deposition of a specific substance in the cerebral cortex. The pathogenesis of AD is commonly linked with the accumulation and aggregation of Aβ and the hyperphosphorylation of tau proteins, leading to neurofibrillary tangles (NFTs) and synaptic disruption [3].
Cognitive and memory impairment in AD is linked to neuroinflammation, oxidative stress (OS), synaptic disruption, and lipid dysregulation [4]. Clinical studies have shown that statin therapy can improve cognitive functions in patients with AD. Additionally, the use of these compounds has been found to reduce the risk of dementia in adulthood. Statins are effective inhibitors of cholesterol synthesis, primarily through the inhibition of HMG-CoA reductase (HMGCR). Statins are known to exert pleiotropic effects, including antioxidant, anti-inflammatory, and neuroprotective impacts, across multiple biological pathways [5]. These properties of statins may underlie their beneficial effects in various pathological conditions, such as AD. These effects will be discussed in detail in this review article.
Statins
Statins are primarily used in the treatment of hypercholesterolemia. These drugs inhibit HMGCR, the rate-limiting enzyme in cholesterol synthesis, which regulates the conversion of HMG-CoA to mevalonic acid. Beyond their cholesterol-reducing effects, statins also lead to the depletion of downstream isoprenoid products of this pathway, including isopentenyl pyrophosphate, farnesyl pyrophosphate, and geranylgeranyl pyrophosphate. This depletion induces changes to small GTPases, such as Ras proteins [6]. These products serve as the primary lipid attachments for the post-translational modification of various proteins, including heterotrimeric G proteins and small GTP-binding proteins of the Ras, Rho, Rap, and Rab GTPase families, as shown in Fig. 1 [7].
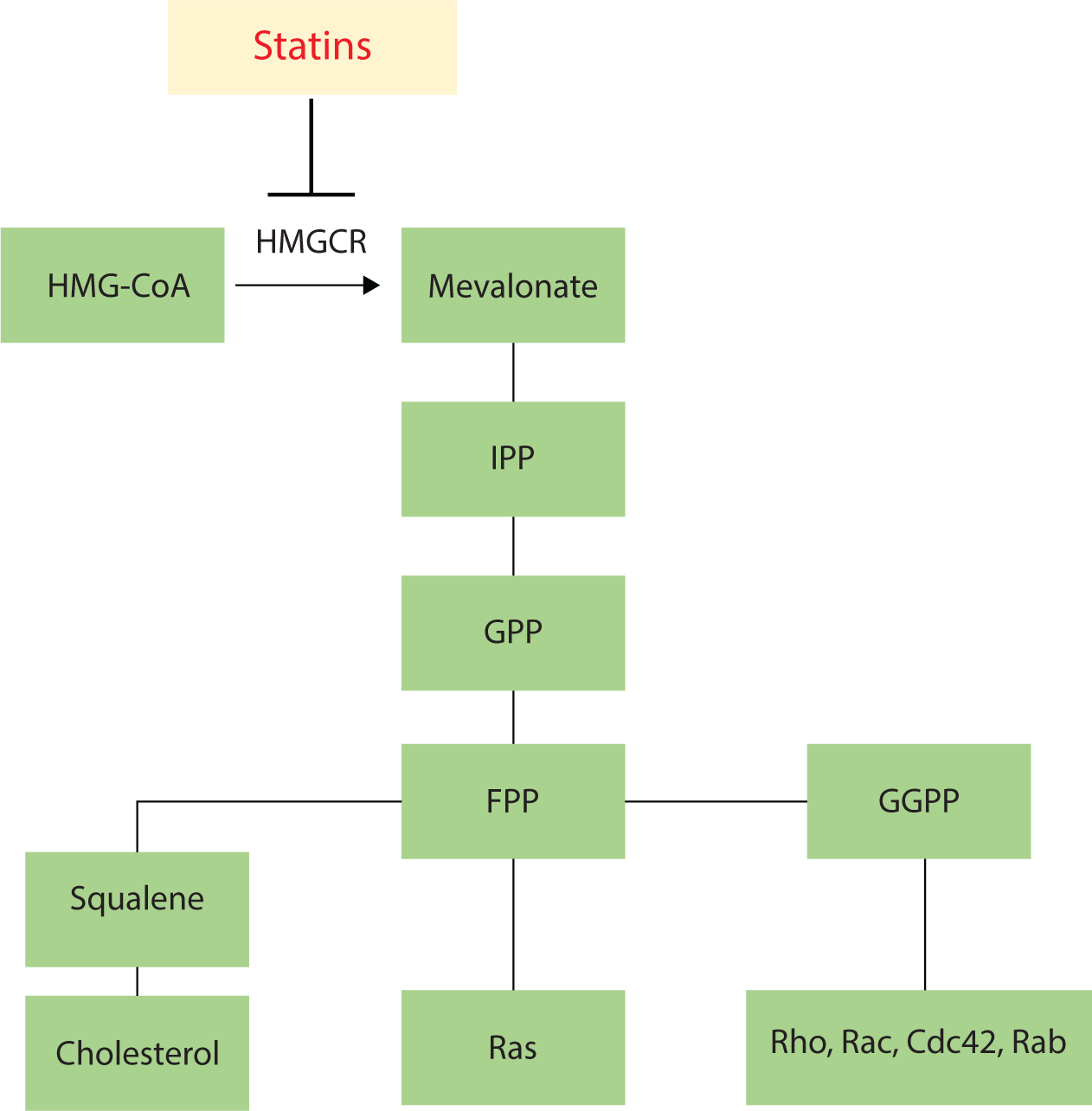
Statins can be categorized into two groups based on their method of production. The first group comprises natural or fungal-derived statins, including mevastatin, lovastatin, pravastatin, and simvastatin. The second group consists of synthetic statins, such as fluvastatin, atorvastatin, cerivastatin, pitavastatin, and rosuvastatin [8]. Statins can also be classified by their solubility into lipophilic and hydrophilic statins. Lovastatin, simvastatin, atorvastatin, and fluvastatin fall under the lipophilic category, while pravastatin and rosuvastatin are classified as hydrophilic (Table 1). The various properties of statins should be considered when utilizing them as effective treatments for brain diseases. Notably, for a drug to impact the brain, it must be able to cross the blood-brain barrier (BBB). Among the statins, lovastatin and simvastatin are the most frequently used in brain research [9].
Lipophilic statins exhibit a greater impact on neurodegenerative diseases and dementia than hydrophilic statins [10]. A study by Pan et al. demonstrated that lipophilic statins were more effective in reducing the risk of AD [11]. However, another study indicated that lipophilic statins were associated with a higher risk of AD compared to hydrophilic statins [12]. These results can be attributed to the superior capacity of lipophilic statins to passively cross the BBB and easily interact with various tissues, including the CNS, adipose tissue, and muscle [13]. In other research, hydrophilic statins have been reported to decrease the risk of AD more strongly (by 28%) than lipophilic statins (16%). In general, hydrophilic statins require a carrier to enter tissues other than the liver [13].
Pathogenesis of Alzheimer Disease
The key agents involved in the pathogenesis of AD have garnered considerable attention, yet the mechanisms of this disorder remain incompletely defined and continue to be questioned. A prevailing theory is the amyloid cascade hypothesis, which proposes that Aβ peptides act as a trigger for subsequent events that lead to AD [14]. AD is marked by the presence of Aβ plaques and NFTs. Aβ peptides spontaneously aggregate into soluble oligomers, fibrils, and senile plaques; these subsequently cause oxidative damage, activation of microglial and astrocytic cells, and changes in kinase/phosphatase activity, ultimately culminating in neuronal cell death [15]. Aβ peptides may also induce tau phosphorylation, which results in the destabilization of microtubules, impaired axonal transport, and eventual neuronal death [16].
Given the evidence that tau hyperphosphorylation can induce neuron death, it is widely recognized that tau-oriented therapeutic strategies could be effective for AD and other disorders characterized by tauopathies [17].
Conversely, it has been determined that Aβ indirectly contributes to neurotoxicity by stimulating microglia to secrete inflammatory substances such as cytokines. Furthermore, an increasing body of evidence suggests that Aβ plaques are associated with local synapse loss and synaptic disruption [18].
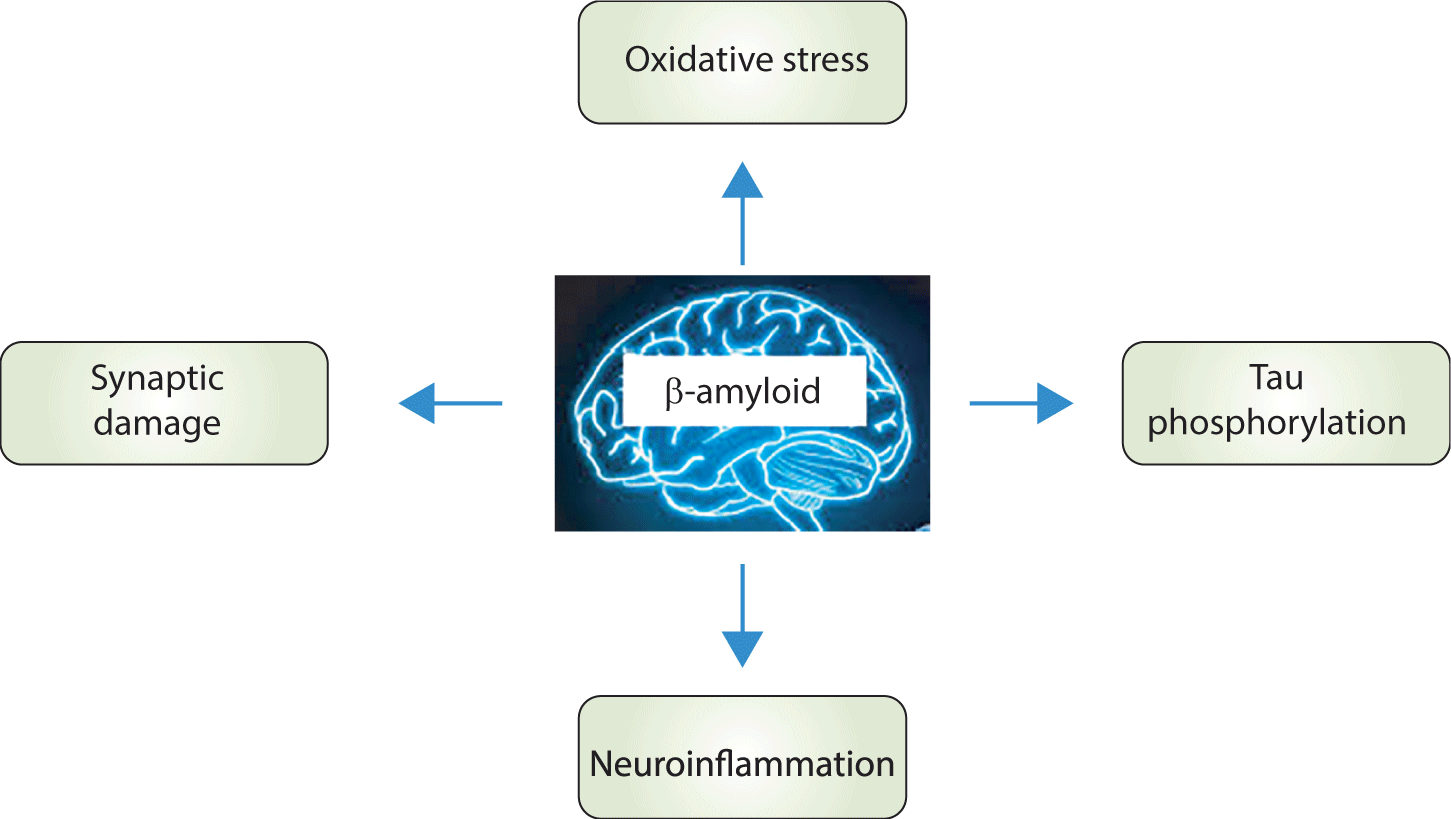
Generally, the memory impairment and dementia observed in AD are associated with the accumulation of Aβ. This accumulation triggers neuroinflammation, increases OS, induces synaptic dysfunction, and promotes tau protein hyperphosphorylation (Fig. 2).
Dysregulation of Lipid Homeostasis and Alzheimer Disease
Evidence from various studies suggests that dysregulation in lipid metabolism homeostasis within the CNS significantly increases the risk of developing AD. Indeed, the concept that maintaining cholesterol homeostasis could offer new potential therapeutic strategies to inhibit or delay the progression of AD is gaining traction [19]. A prior study indicated that midlife cholesterol levels were elevated in individuals exhibiting mild memory loss or dementia [20]. Similarly, high cholesterol levels at midlife have been found to significantly heighten the risk of late-life dementia [21]. Additionally, aging is a key risk factor for developing some forms of dementia, such as AD. In humans, aging is associated with increases in plasma concentrations of triglycerides, cholesterol, and LDL. High cholesterol levels are associated with an elevated risk of memory loss and AD. This implies that the risk of developing neurodegenerative diseases such as dementia and AD is dramatically increased in older adults [22].
Cholesterol levels in the cortical gray matter of the brains of patients with AD have been found to be elevated by 19% to 34% [23]. A diet high in cholesterol can approximately double the levels of Aβ in the hippocampus, leading to damage to the BBB [24]. Dysregulation of lipid homeostasis may elevate Aβ levels by influencing the cleavage of amyloid precursor protein (APP), a primary risk factor associated with the pathogenesis of AD [25].
A substantial body of research on the potential link between high cholesterol levels and increased AD risk suggests that reducing cholesterol levels could be an effective strategy for the treatment or prevention of AD. A growing amount of evidence suggests that chronic treatment with statins, as HMGCR inhibitors, reduces the risk of developing AD. Statins can reduce the formation of Aβ by lowering cholesterol levels. Initial results supporting the favorable effects of statins in AD have been obtained from two studies; the findings indicated that these drugs were associated with up to a 70% reduction in AD incidence [26,27]. Supporting the previous notion that statins can have beneficial impacts on neurocognitive disorders, a study by Wolozin et al. found that lovastatin and pravastatin were associated with a reduced risk of AD development [28]. Additionally, the capacity of statins to combat AD may stem from the prevention of protein isoprenylation [29]. Aβ, the primary component of senile plaques in AD, is a product derived from APP. The knockout of Rho family protein activity reduces levels of APP C-terminal fragments by enhancing lysosome-dependent degradation. A previous study indicated that statins may reduce Aβ formation by inhibiting proteins of the Rho and Rab families, suggesting a mechanism of statin action in AD [30]. This research proposes an additional potential mechanism by which statins can mitigate AD pathogenesis, via the inhibition of protein isoprenylation.
Protective Effects of Statins
The accumulation of high levels of free radicals, defined as OS, is hazardous. This accumulation may result from either excessive production or insufficient removal of radicals. The removal process can occur through several potential mechanisms and is typically carried out by an antioxidant compound. These compounds are molecules that substantially delay or prevent the oxidation of an oxidizable substrate. Antioxidant mechanisms include the scavenging of free radicals, the quenching of sources of free radicals, and the regeneration of endogenous antioxidants [31].
The reactive oxygen species (ROS) family of compounds includes the partially reduced oxygen species O2•–, H2O2, and HO•. These free radicals are generally defined as oxygen-containing chemicals that possess reactive properties. An increased level of ROS within a biological system can interact with fundamental biological macromolecules such as DNA, RNA, lipids, and proteins. The brain is particularly vulnerable to oxidative damage induced by OS, given its substantial consumption of dioxygen and its high lipid content [32].
OS manifests early in the progression of AD, reinforcing its potential role in the pathogenesis of this disease, particularly in relation to the presence of Aβ. In fact, elevated levels of Aβ correlate with an increase in oxidation compounds derived from proteins, lipids, and nucleic acids in the hippocampus and cerebral cortex [33].
Research has established that Aβ impairs mitochondrial redox activity and increases the production of ROS. Various studies provide evidence that Aβ-induced OS results in the apoptotic death of neuronal cells, a process that can be inhibited by antioxidants [34]. These data imply that ROS could serve as a crucial mediator of Aβ-induced neuronal cell death in the progression of AD.
The proposed association between OS and elevated AD risk suggests that antioxidant compounds could be a suitable treatment for AD. The antioxidant properties of statins are recognized as an inherent characteristic of these drugs, first documented in vitro [35]. In an investigation of the antioxidant activity of statins, Franzoni et al. found that all of these compounds exhibited antioxidant effects, but they varied in potency and the types of free radicals most effectively scavenged [36]. More specifically, research has indicated that fluvastatin demonstrates the greatest anti-peroxyl radical antioxidant effect, while simvastatin shows the highest anti-hydroxyl radical activity [37]. Thus, the antioxidant effects of statins may vary depending on the targeted substrate.
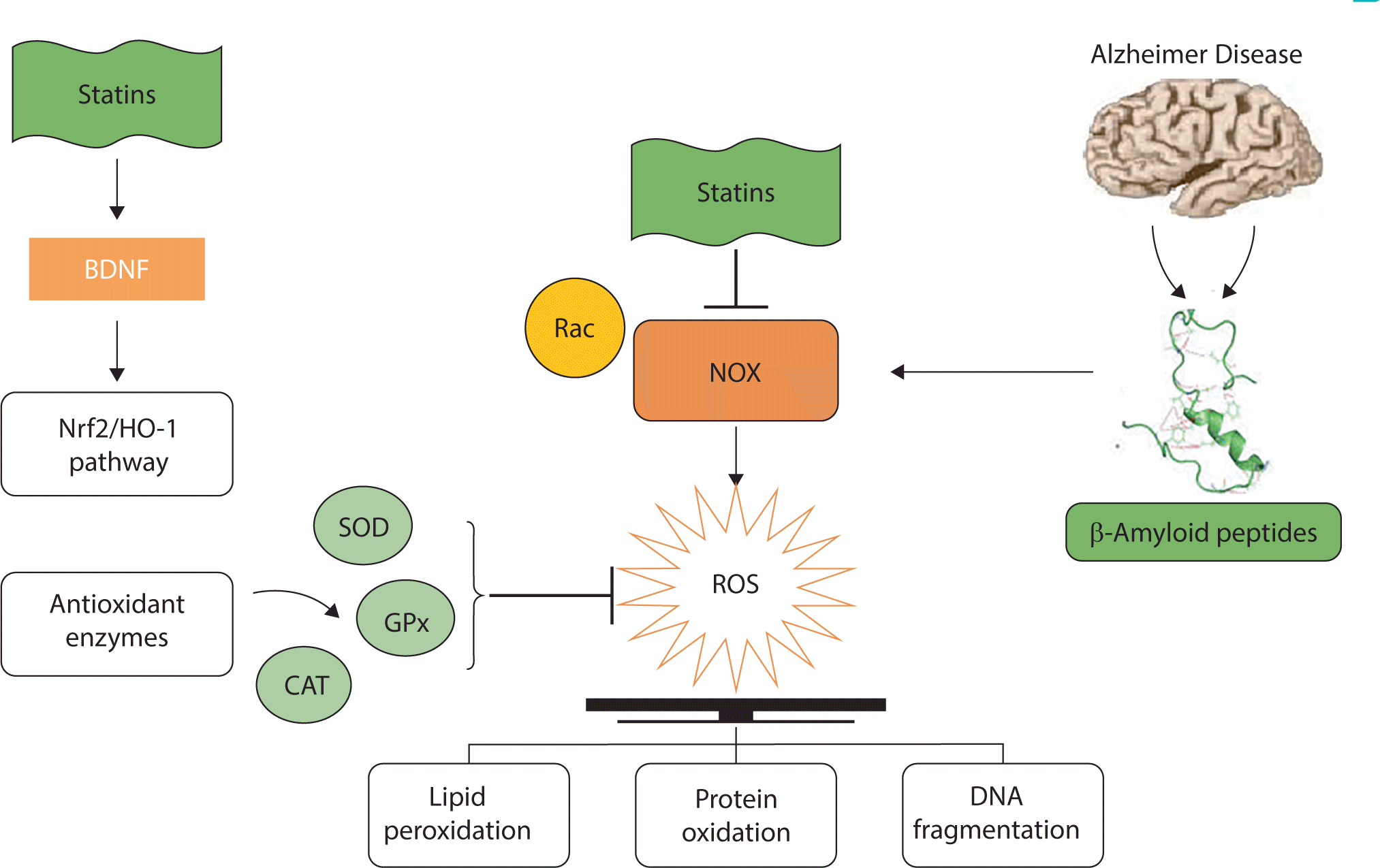
The accumulation of Aβ in the brain is a crucial pathogenic event in AD. Among the various mechanisms proposed to explain the neurotoxicity of Aβ deposits, one suggests that Aβ indirectly causes neurotoxicity by activating microglia to produce ROS. Nicotinamide adenine dinucleotide phosphate oxidase (NOX) is a key contributor to oxidative damage and processes. Increased NOX activity is known to lead to the development of various diseases by producing excess ROS and establishing OS conditions [38]. Consequently, inhibiting this enzyme could be a promising therapeutic strategy for the treatment of various diseases. Aβ engages with microglia, activating these cells and causing the overproduction of O2•– by NOX. This O2•– is then converted into H2O2 and ultimately to •OH in the Fenton reaction. Aβ peptides induce mitochondrial dysfunction and OS through the activation of NOX [39]. Statins are well-known to prevent the toxic effects of oxidative conditions by suppressing the activation of the NOX pathway [40]. Specially, Rac plays a key role in the activation of NOX and the subsequent production of ROS [41]. Interestingly, statins inhibit Rac activity and effectively prevent protein interaction and NOX production, ultimately reducing ROS generation.
Additionally, research has demonstrated that statins upregulate the activity of antioxidant enzymes such as glutathione peroxidase, catalase, and superoxide dismutase, which eliminate ROS while generating oxygen and water [42]. A pathway involving nuclear factor erythroid 2-related factor 2 (Nrf2), a key transcription factor, can regulate the expression of certain genes. The protein products of these genes contribute to the detoxification and elimination of ROS through conjugative reactions and by increasing cellular antioxidant capacity. Furthermore, Nrf2 is a crucial factor in regulating the expression of endogenous antioxidants, including heme oxygenase-1 (HO-1). The Nrf2/HO-1 signaling pathway is reportedly instrumental in cellular responses under oxidative conditions [43]. Nrf2 elevates the expression levels of genes related to antioxidant enzymes and promotes the synthesis of these enzymes in astrocytes. Notably, antioxidant enzymes produced in astrocytes are transported to neuronal cells, providing a protective effect against OS [44]. Statins have been found to influence the Nrf2/HO-1 signaling pathway, thereby safeguarding cells against the destructive effects of ROS [45]. Habeos et al. reported that statins could augment the DNA-binding activity of Nrf2, leading to the expression of its target genes, including HO-1 and glutathione peroxidase, and protecting cells from ROS toxicity [46]. Increased lipid peroxidation is a common risk factor for AD, as it diminishes membrane integrity and alters the permeability of various ions in the plasma membrane. Studies have shown that statins can scavenge ROS and markedly reduce lipid peroxidation [47,48].
Overall, the beneficial effects of statins against oxidative damage are indisputable and have been confirmed in multiple neuropathological conditions, ranging from brain injuries to AD. Statins reduce OS by stimulating the Nrf2/HO-1 pathway and inhibiting the NOX pathway (Fig. 3).
Inflammation is a response that occurs in reaction to injury, trauma, or infection affecting cells and tissues. A substantial body of evidence in the literature details the biochemical connection between the brain and the immune system. Inflammation occurring within the brain is identified as neuroinflammation. Microglia and astrocytes, two key components of the brain’s immune system, play pivotal roles in the process of neuroinflammation [49].
Inflammation is a primary contributor to the development of neurological disorders, as evidenced by the heightened expression of inflammatory factors in the CNS. Findings from the 1980s indicate that immune-related factors are present near Aβ plaques [50]. It is now established that the release of cytokines and the broad activation of associated receptors in the brains of patients with AD lead to neuroinflammation [51]. However, some studies have demonstrated that a persistent immune response in the CNS is linked to neurodegeneration and can also induce and exacerbate Aβ pathologies [52]. Furthermore, inflammation may mediate a connection between early Aβ pathology and the subsequent development of NFTs [53].
Activation of microglial cells triggers the generation and release of proinflammatory cytokines. These cytokines stimulate neighboring astrocytes to further produce Aβ oligomers, which may stimulate neuronal cell death by intensifying inflammation [51]. In AD, the accumulation of intraneuronal Aβ, which forms senile plaques, instigates neuroinflammation through microglial activation. This process results in neurodegeneration and ultimately escalates neuronal death in the hippocampus. Given the clear link between statin administration and the reduction of inflammatory responses in AD, decreased neuroinflammation could be proposed as a key mechanism for the neuroprotective effects of statins. In AD, a reduction in Aβ formation has been associated with decreased neuroinflammatory responses [54]. One study demonstrated that atorvastatin prevents Aβ-induced microglial activation, which marks the initial phase of neuroinflammation [55].
The activation of microglia results in the release of several inflammatory factors, including TNF-α, IL-1β, and IL-6. These factors can exacerbate the pathogenesis of neurodegenerative disorders by establishing an inflammatory microenvironment. The secretion of TNF-α and IL-6, both proinflammatory mediators, promotes neuronal apoptosis [56]. IL-6 is primarily synthesized in neuroglia, and it triggers the accumulation of inflammatory cells and the increased production of ROS during inflammation; furthermore, it collaborates with TNF-α to induce calcium overload and cell apoptosis [57]. Consequently, TNF-α is considered an effective target for the treatment of inflammatory diseases. A connection between statins and TNF-α production has been established. Previous research has indicated that statins can reduce the production of TNF-α and interferon gamma [58]. Lindberg et al. found that microglial cultures exposed to statins (specifically, atorvastatin and simvastatin) demonstrated a decrease in IL-6 levels [59]. Separately, Inoue et al. suggested that the cytokine inhibitory effects of statins on the capacity of IL-6 to promote inflammation may be linked to interference with the phosphorylation of the transcription factor STAT3 [60].
Beyond TNF-α and IL-6, the nuclear transcription factor kappa B (NF-κB) plays an important role in pathological processes and the generation of pro-inflammatory cytokines. NF-κB is instrumental in activating and advancing key events in AD, such as CNS dysfunction and neuroinflammation. Encouraging research has been published on the modulation of NF-κB activation within the CNS, which subsequently attenuates the processes that trigger neural degeneration [61].
As reported by Hilgendorff et al., all statins including atorvastatin, cerivastatin, fluvastatin, lovastatin, pravastatin, and simvastatin inhibit NF-кB activity in monocytes. Despite this common property among statins, notable differences exist in the degree of inhibition provided. The potency of the statins, ranked from highest to lowest, is as follows: cerivastatin, atorvastatin, simvastatin, pravastatin, lovastatin, and fluvastatin. These differences are also reflected at the transcriptional level and the protein level of NF-кB–regulated tissue factor expression [62]. In a separate study conducted by Hernandez-Presa et al., treatment with simvastatin in a rabbit model of atherosclerosis yielded a decrease in the activity of the transcription factor NF-κB [63].
In the context of neuroinflammation, NF-κB regulates several genes, including TNF-α, IL-1β, and IL-6, through the nuclear translocation of transcription factors [64]. Monica et al. conducted a study to investigate the impact of statins on NF-κB activity, which is known to induce the messenger RNA expression of chemokines. Their findings suggest that statins can reduce inflammation by inhibiting both NF-kB activity and chemokine gene expression [65].
The enzyme cyclooxygenase (COX) has been documented to be important for the synthesis of prostaglandins, which are potent inducers of inflammation. Certain treatments have demonstrated positive effects in inhibiting neuroinflammation associated with disease progression through the inhibition of COX. Research has also indicated that the expression of COX-2 is primarily regulated by NF-κB [66]. Given that several studies affirm the role of NF-κB in COX-2 expression, inhibiting NF-κB can suppress COX-2 expression. Atorvastatin has been shown to mitigate inflammation by reducing the activity of NF-κB, in turn decreasing the expression of the pro-inflammatory enzyme COX-2 [63]. Ramalho et al. reported that the inactivation of NF-κB can inhibit the expression and synthesis of certain pro-inflammatory agents, including IL-6, TNF-α, and COX-2, as well as prostaglandin E2 synthesis [67]. Therefore, based on the studies conducted, the inflammatory response appears to be an important characteristic of the pathogenesis of AD. Aβ stimulates inflammatory responses by activating NF-κB, which subsequently produces cytokines such as IL-1β, IL-6, TNF-α, and COX-2 [65]. Thus, inhibiting the production of these factors could be a potential therapeutic strategy for AD. Indeed, statins have been shown to prevent Aβ-induced microglial activation, an initial phase in neuroinflammation (Fig. 4).

Long-term potentiation (LTP), a form of synaptic plasticity, has been identified as the primary mechanism involved in the plasticity related to learning and memory [68]. Two recognized types of LTP occur in different regions of the mammalian nervous system. The first type relies on the activation of the N-methyl-D-aspartate receptor (NMDAR), while the second type, which has been less extensively studied, is NMDAR-independent. The most prevalent form of LTP involves the activation of the glutamate ionotropic NMDAR, which is highly permeable to calcium ions [69].
Early functional and structural synaptic loss in AD correlates with the severity of the disease. Multiple studies have revealed the mediating role of Aβ in the key mechanism involved in LTP impairment [70,71]. In the initial stages, Aβ oligomers induce reversible synaptic alterations that largely mirror those involved in normal action-dependent synaptic plasticity and dendritic pruning during development in the adult brain. However, in the later stages of the disease, tau-related neuropathology triggers irreversible changes [72]. Research from animal models suggests that Aβ interacts with tau to induce synaptic degeneration [17]. One important finding is that Aβ inhibits the induction of LTP in the hippocampus, specifically in the CA1 and DG areas [70,73]. Chen et al. reported that Aβ particularly impacts both presynaptic and postsynaptic neurons, reducing the NMDA peak amplitude [71]. Previous studies have indicated that statins can reduce Aβ levels in the CNS [74,75]. Moreover, atorvastatin has been confirmed to decrease Aβ by reducing APP, β-secretase, and OS in clinical models of dementia [76,77]. Simvastatin has also been reported to enhance vascular activity in the brain, decrease neuroglia activation, and reduce the number of dystrophic neurites induced by Aβ plaques [78].
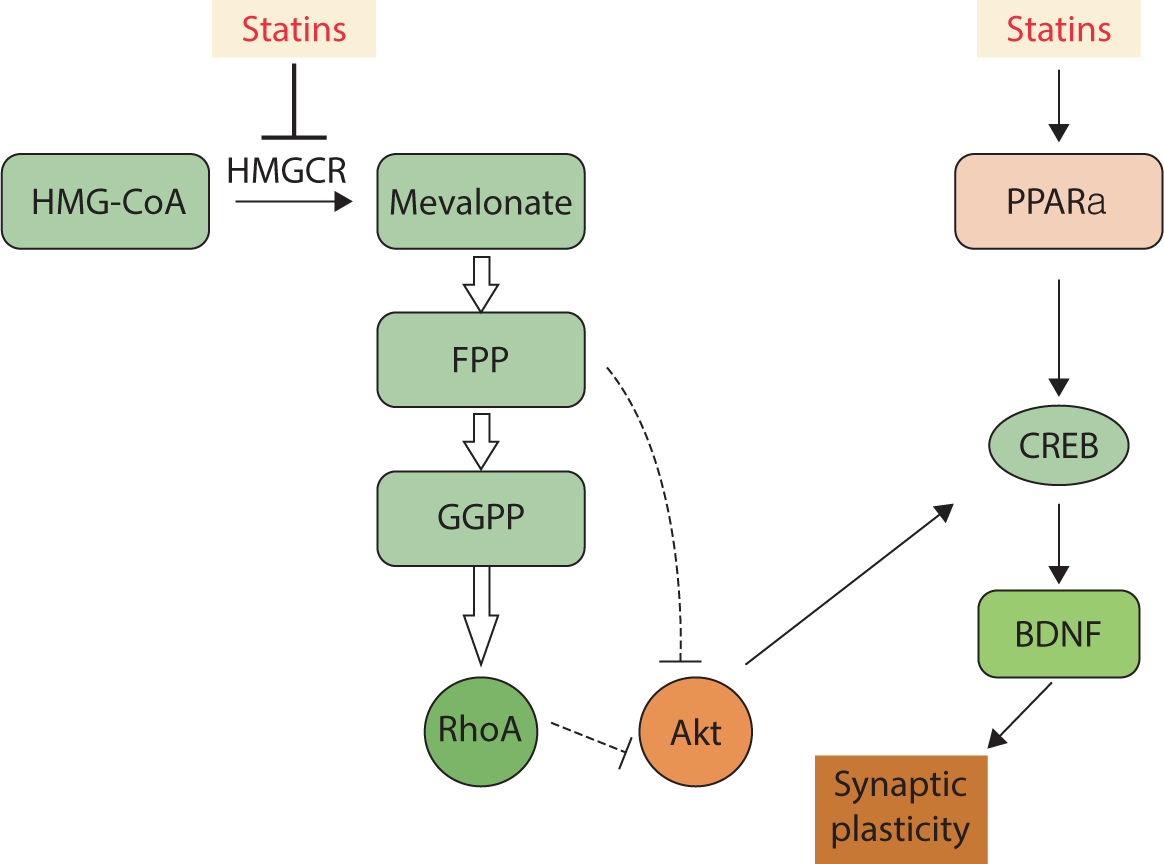
Research supports the involvement of the Rho/Akt/cAMP response element-binding protein (CREB) pathway in the enhancement of LTP and memory induced by statins. This indicates that statins promote the upregulation of the memory-related genes encoding c-Fos and Egr-1. The transcription of these genes takes place downstream of CREB activation [79]. Experiments conducted on the hippocampal CA1 region have shown that statins can enhance LTP by inducing Akt phosphorylation [80]. Besides directly inhibiting the production of mevalonate, statins influence synaptic plasticity and memory function through mechanisms that are independent of the mevalonate pathway. As ligands for peroxisome proliferator-activated receptor alpha, statins increase the transcription of CREB, thereby inducing the expression of neurotrophic factors such as brain-derived neurotrophic factor and neurotrophin-3 [81] (Fig. 5).
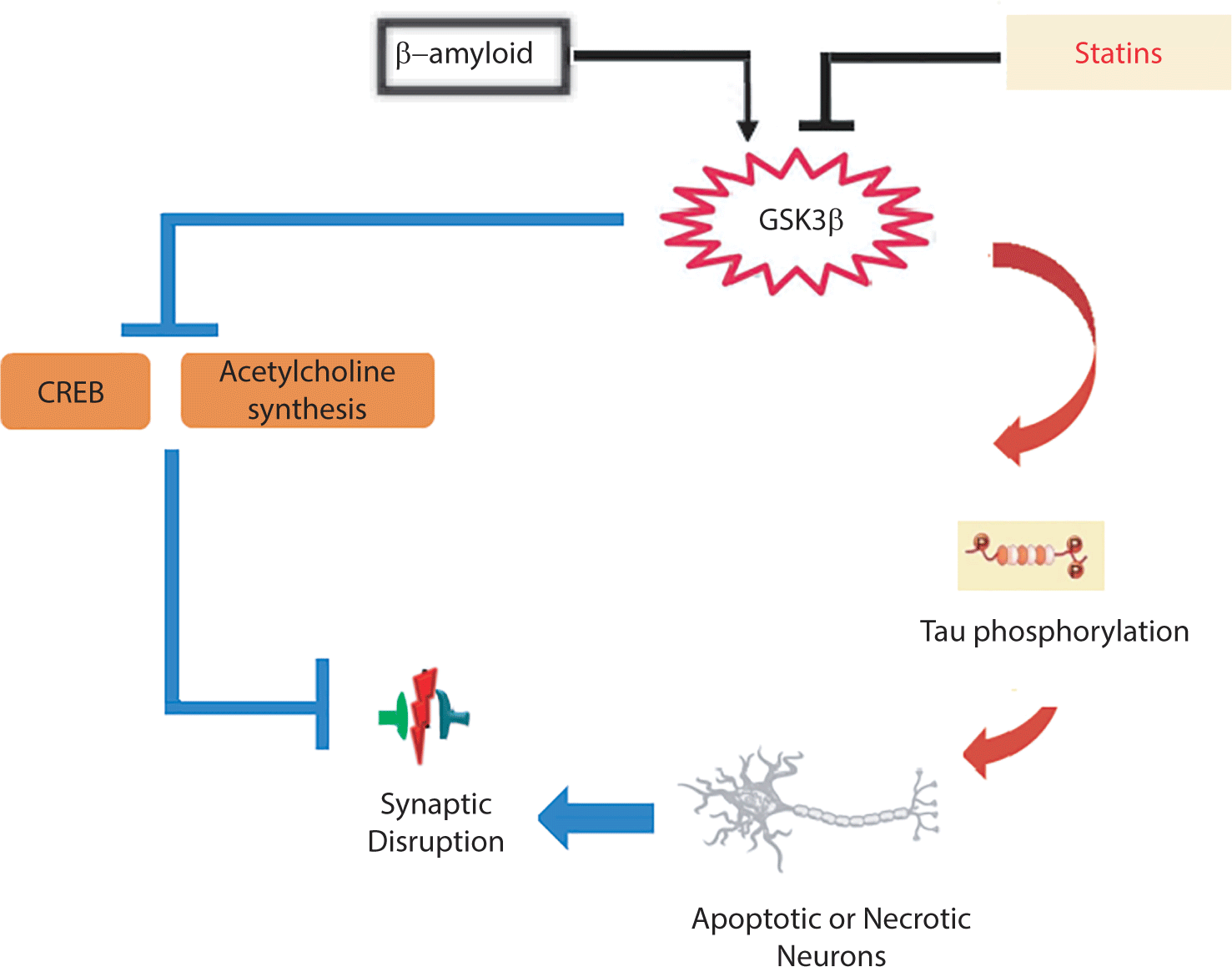
Glycogen synthase kinase 3 (GSK3) is a crucial enzyme in glycogen metabolism, with two isoforms: GSK-3α and GSK-3β. Research has shown that GSK-3β is associated with synaptic dysfunction and memory disruption. The activation of this enzyme results in a deficiency in LTP and an induction of long-term depression through its impact on NMDA receptors [82]. GSK-3β activity has been shown to lead to tau hyperphosphorylation, which can intensify neurodegeneration, synaptic dysfunction, and neuronal death in the hippocampus [83]. In other words, Aβ triggers abnormal activation of GSK-3β, resulting in the loss of dendritic spines and changes in spine morphology, which can contribute to neuronal loss in AD [84]. Interestingly, statins have been found to decrease the activity of GSK-3β, which is responsible for tau protein phosphorylation. As reported by Salins et al., statins lower the production of senile plaques by reducing the activity of GSK-3β, ultimately protecting neurons from the toxicity caused by Aβ [85].
AD and dementia strongly impact cholinergic neurons, which play an important role in memory function. Two types of receptors for acetylcholine (ACh) include nicotinic receptors (nAChRs) and muscarinic receptors (mAChRs). The selective loss of α7 nAChRs in the brains of patients with AD suggests a connection between this nAChR subtype and the pathophysiology of the disease [86]. In general, two potential strategies are used to address the cholinergic system dysfunction observed in patients with AD and dementia. One strategy involves the use of acetylcholinesterase inhibitors, which prevent the breakdown or hydrolysis of ACh, thereby increasing its levels in the CNS [87]. The second strategy proposes the activation of nAChRs and mAChRs to restore cholinergic function in patients with AD [88]. Zhao et al. reported that lovastatin can significantly increase the activity of choline acetyltransferase in the cortex and hippocampus [89]. In contrast, simvastatin reduces acetylcholinesterase activity in the frontal cortex of rats, which may consequently increase the levels of ACh in the synaptic cleft and alleviate the cholinergic dysfunction in AD [90]. GSK3 has also been established to decrease ACh synthesis, a process associated with the cholinergic deficiency in AD [91]. Interestingly, it has been previously noted that statins can decrease the activity of GSK-3β [85].
Overall, studies indicate that the activation of GSK-3β, followed by tau hyperphosphorylation, CREB inhibition, and cholinergic system deficiency, contribute to neuronal loss in patients with AD. Statins appear capable of enhancing synaptic plasticity by inhibiting the activation of GSK-3β (Fig. 6).
Conclusion
AD is a devastating age-related neurodegenerative disease with a rapidly increasing prevalence. In addition to Aβ and NFT plaques, the brains of patients with AD exhibit signs of a persistent oxidative state, neuroinflammatory response, and synaptic disruption. Aβ stimulates microglial proliferation and triggers the release of ROS due to NOX activation. Extensive research suggests that the role of NOX in the development of various diseases, through excessive production of ROS and establishment of an OS condition, implies that the inhibition of this enzyme by statins is responsible for statin-induced antioxidant effects. Additionally, statins exert antioxidant effects through the stimulation of the brain-derived neurotrophic factor/Nrf2 pathway and the subsequent reduction in intracellular ROS via the preservation and enhancement of endogenous antioxidants. Neuroinflammation is another key factor involved in the progression of AD, as evidenced by the increased release of inflammatory factors in the CNS. In AD, the interaction of Aβ fibrils with microglial cells leads to the activation of NF-κB, which in turn promotes the release of inflammatory cytokines including IL-1β, IL-6, TNF-α, and COX-2. Statins can reduce neuroinflammation by inhibiting NF-κB transcription factor activity and subsequently suppressing the release of inflammatory cytokines. According to the findings, early functional and structural synaptic loss in AD correlates with the severity of the disease. An aberrant increase in GSK-3β activity, induced by Aβ in the CNS, results in the loss of dendritic spines, morphological changes of spines, and neuronal loss. GSK-3β activation and subsequent tau hyperphosphorylation, CREB inhibition, and cholinergic system deficiency are implicated in synaptic disruption in AD patients. Statins appear to improve synaptic plasticity through the inhibition of GSK-3β activation. Given that statins exhibit several beneficial properties, including neuroprotective, anti-inflammatory, and antioxidant effects, they may be effective in treating neurological diseases.